
Bravais lattices and unit cells are an efficient way of describing a periodic structure. Another way of looking at a structure is by counting the number of nearest neighbours around each atom and the geometry of their distribution. In general, this will be different for each distinct point in the unit cell. This approach is particularly useful in non-crystalline structures, where a description based on a periodic lattice isn't adequate. The number of nearest neighbours is the coordination number and the geometric shape resulting from the arrangement of the neighbours is the coordination polyhedron, with the atom under consideration at its centre.
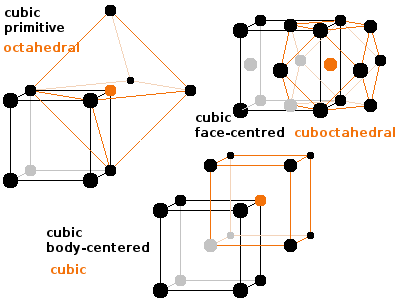
Comparing the three cubic Bravais lattices shows that each atom in the primitive cubic (or simple cubic (sc)) lattice has six neighbours at distance $$s=a$$ (the lattice constant of the cubic unit cell), arranged as an octahedron. The coordination polyhedron of the body-centred cubic (bcc) lattice is a cube of the same size and orientation of the unit cell. In other words, the bcc lattice consists of two identical inter-penetrating cubic lattices offset by half a spatial diagonal $$s=\frac{\sqrt{3}}{2}a\qquad;$$ the centred atom of one lattice is a corner atom in the other, and vice versa. The coordination number is eight. Each atom in the face-centred cubic (fcc) structure has twelve equidistant neighbours at a distance of $$s=\frac{1}{\sqrt{2}}a\qquad,$$ half a face diagonal, in the corners of a cuboctahedron.
Different crystals can have the same structure type. This means the atoms are located in the same places within the unit cell and the symmetry is the same. Of course, the atoms and ions of different elements are different size, which results in different bond lengths, so the dimensions of the unit cell will be different. For example rock salt, Na+Cl-, and magnesium oxide, Mg2+O2-, share the same structure type. For different crystals to have the same structure type, the ratio of the individual elements in the formula unit needs to be the same (or we wouldn't be able to replace atoms in the unit cell one-by-one) and the size ratio of the atoms or ions must be similar (or we would leave gaps, and another, denser structure would be more stable).
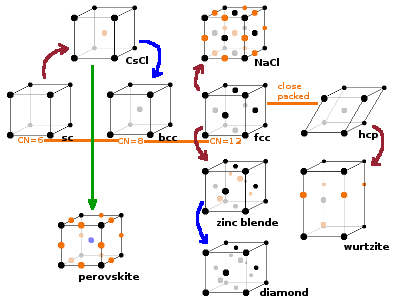
Most structure types can be related to each other by:-
Within a given crystal lattice, different structure types are distinguished by the coordination number of their atoms. For example compare the number of neighbours of each atom in simple, body-centred and face-centred cubic structures. Another way of looking at the structures is by representing each atom by a hard sphere, which gives rise to the two close packed structure types.
Some of the more common structure types are presented below in more detail. This list is far from exhaustive, though! Each panel shows a representation of the unit cell (left), the coordination polyhedra of each atom type (right), and slices through the unit cell at several heights (bottom). Most have a link to a Flash applet on an external site, which allows rotating and zooming into the structures as well as looking at unit cells from different angles.
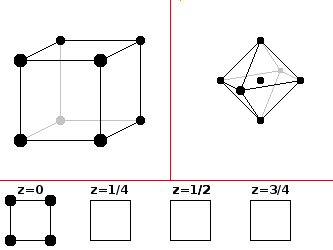
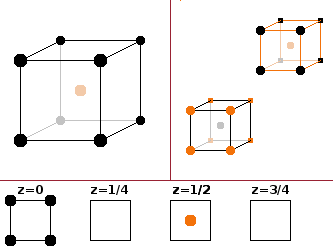
The simple cubic (sc) type is just a primitive cubic unit cell with one atom type and one atom per cell (in the corner, repeated in all corners because the corners are shared with the neighbouring seven cells). The coordination polyhedron is an octahedron - six equidistant neighbours, of which four are in a plane and the other two above and below the central atom. This type is relatively rare because it leaves a large gap at the centre of the unit cell, and it is usually energetically beneficial for the crystal to reconfigure into one of the close-packed structures. Polonium is the only element crystallising in this form.
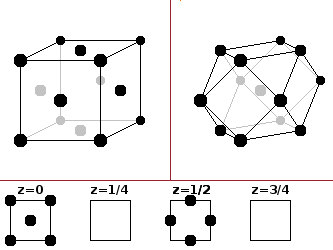
The face-centred cubic (fcc) structure derives from the simple cubic type by adding an additional atom in the centres of all the faces of the cubic unit cell. Since faces are shared between two neighbouring cells, the total number of atoms in the unit cell is four.
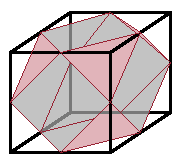
The coordination polyhedron is a
cuboctahedron,
i.e. a cube whose corners have been cut off,
resulting in a coordination number of 12.
The basis is: 0 0 0 -- ½ ½ 0 -- 0 ½ ½ -- ½ 0 ½.
Elemental metals often crystallise in this structure.
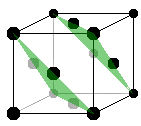
The fcc type is also known as cubic close-packed (ccp), because it can be visualised as layers of hard spheres packed densely and stacked along the spatial diagonal of the cube. The picture shows how the close-packed lattice planes, spanned by three corner atoms each, intersect the cubic unit cell. More on close-packed structures below.
The caesium chloride type is characteristic of binary compounds and alloys where the two species are very similar in size, resulting in two identical sub-lattices. The type derives from the simple cubic structure by interlacing the two identical simple cubic sub-lattices shifted by half a spatial diagonal. Each atom type occupies one of the sublattices, and the cell contains one atom of each type (Cl- at 0 0 0, Cs+ at ½ ½ ½), and the coordination polyhedron for each looks like the unit cell itself: a cube (coordination number 8) with an atom of the opposite type in each corner.
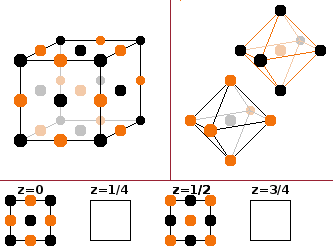
The
rock salt type
occurs in fully
ionised binary compounds
where there is a
large size difference
between cations and anions. Each atom type is in its own fcc lattice; the
two identical fcc sub-lattices
are offset by half a spatial diagonal. Each
ion is coordinated by six ions of the other species forming an
octahedron.
The basis includes eight atoms, bearing in
mind that corner atoms are shared between eight, edge atoms between four, and face atoms between two cells:
Na+: ½ ½ ½ -- ½ 0 0 -- 0 ½ 0 -- 0 0 ½
Cl-: 0 0 0 -- ½ ½ 0 -- 0 ½ ½ -- ½ 0 ½
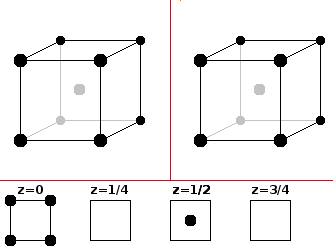
The body-centred cubic (bcc) structure is less dense than the fcc type, but denser than a simple cubic arrangement. The alkali metals and tungsten crystallise according to this pattern. Because of the centred atom, the coordination polyhedron is a cube - identical to the unit cell itself. The basis is 0 0 0 -- ½ ½ ½. It is identical to the caesium chloride structure if both positions are occupied by the same atom type.
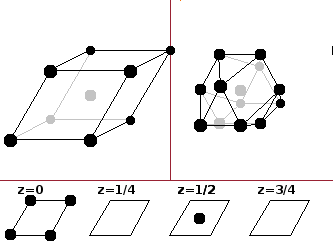
The
hexagonal close-packed (hcp)
type is, like the
fcc type,
made up of layers of hard spheres arranged in a hexagonal pattern. For the difference between the two, see the section on
close-packed
structures below. The coordination number again
is twelve, this time arranged in the shape of an
anti-cuboctahedron,
i.e. a cuboctahedron that has been twisted by 45 degrees
around its central plane, such that the top and bottom square surfaces are askew. The basis is 0 0 0 -- ½ ½ ½.
The hcp type is common among elemental metals.
The perspective of the diagram may be a little misleading: The z-slices are vertical
cross-sections through the cell. The base of the cell viewed from this angle is square (but distorted due to the perspective)
and the front and side faces are diamond-shaped.
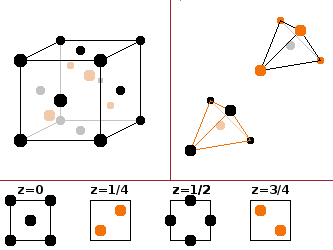
Zinc blende and wurtzite are two forms of the mineral zinc sulphide, ZnS. The zinc blende structure derives from the fcc structure by stacking two fcc unit cells into one another, offset by a quarter of a spatial diagonal. One of these identical sub-lattices is populated by cations (zinc in the case of the type mineral), the other by anions (sulphur). This results in tetrahedral coordination for both species, but for both only every other tetrahedral gap is occupied by an atom of the opposite species, the remaining ones being left empty. This structure is common among many IV-IV and III-V compounds (ones that are made up of two elements of the 4th or one from the 3rd and one from the 5th main group of the periodic table). Important examples are the semiconductors InSb, ZnSe, AlP as well as SiC.
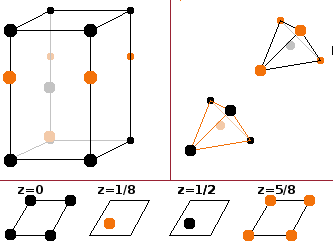
The Wurtzite type derives from the hcp structure in the same way the zinc blende structure derives from the fcc type; it is a matter of stacking order. If layers of zinc and sulphur atoms are denoted by western and Greek letters, respectively, the Wurtzite structure has the hexagonal stacking sequence ...AαBβ... while zinc blende is based on the cubic ...AαBβCγ... pattern. See below for stacking sequences.
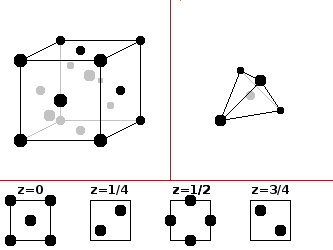
The diamond type is fairly unique to diamond itself. It derives from the zinc blende structure by placing the same atom type in both the corner/face and interior positions. This results in a very dense structure, which is only suitable for very small atoms such as carbon. The individual atoms are tetrahedrally coordinated.
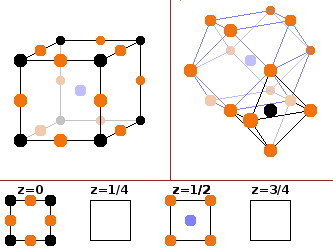
The perovskite type is one of a number of common structures of ternary compounds. The type mineral has the composition CaTiO3, but the structure type is common for other ternary oxides with the formula ABO3. Many of these have important technological applications because they tend to be oxygen vacancy conductors - this means defects where an oxygen atom is missing in the structure are highly mobile and can be used to transport oxygen through the material. This is useful for fuel cells and oxygen sensors. The A atom (green) at the centre of the unit cell has twelve nearest neighbours (all of the oxygens), arranged in a cuboctahedron. The B atom takes the corners of the unit cell, resulting in an octahedral coordination with oxygen. Together, the two cation species make up a bcc lattice, and oxygen atoms are placed in the centres of all edges of the unit cell.
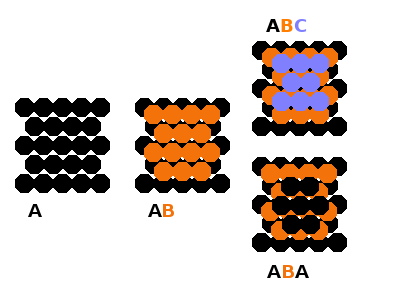
Close-packed structures are visualised as layers of hard spheres packed to maximum density both within layers and between adjacent layers. For a single layer, this is a hexagonal arrangement of neighbours around a central sphere which touch each other and the central sphere.
The spheres of the next layer are placed in the gaps formed by three touching spheres, but only every other such gap can be filled because the distance between them is only half the diameter of the spheres. This defines the second (B, orange) layer. For the third layer, there are two possibilities: the spheres can take the positions corresponding to the unoccupied gaps in the B layer (creating a C layer, green), or the spheres can go in the same positions as in the A layer at the bottom.
With three distinct positions, we can have regular stacking patterns of ...ABCABC... or ...ABABAB... . These are known as the cubic (ABC) and hexagonal (AB) close-packed structure types because the corresponding unit cells are cubic and hexagonal, respectively. Of course, with the same hexagonal pattern within each layer, both structures could be described using a hexagonal Bravais lattice, but for the ABC variant, a more compact unit cell can be defined by stacking the layers along the spatial diagonal of a cube, which corresponds to the fcc structure type.
Energetically, there isn't much difference between either stacking pattern, although the activation energy for moving a whole layer from one position to another is huge. Stacking faults are common, and the two structures have to be regarded as end points of a series of polytypes with varying degree of disorder in stacking sequence.
Now we know a few common structure types, it is time to find out how we can determine crystal structure experimentally using diffraction techniques.