
The Schrödinger equation for an atom with two electrons contains two kinetic energy terms (one for each electron) and three potential energy terms (one each for the Coulomb attraction of each electron by the nucleus, and one mutual repulsion term of the two electrons):
$$\left(\underbrace{\bbox[lightblue]{-\frac{\hbar^2}{2m}\hat{\nabla}_1^2}\bbox[lightgreen]{-\frac{\hbar^2}{2m}\hat{\nabla}_2^2}}_{\textrm{kinetic}}\underbrace{\bbox[lightblue]{-\frac{Ze^2}{4\pi\varepsilon_0r_1}}\bbox[lightgreen]{-\frac{Ze^2}{4\pi\varepsilon_0r_2}}\bbox[yellow]{+\frac{e^2}{4\pi\varepsilon_0r_{12}}}}_{\textrm{potential}}\right)\psi=E\psi\quad.$$
Here, $r_{12}$ is the distance between the two electrons. The two blue terms together are the normal Hamiltonian for a hydrogen-like atom (with the appropriate $Z$ of course); the same applies to the two green terms. If it weren't for the yellow repulsion term, the Schrödinger equation would essentially be the same as for the hydrogen-like atom: $$\left(\bbox[lightblue]{\hat{H}_1}+\bbox[lightgreen]{\hat{H}_2}\right)\psi=E^{(0)}\psi\quad,$$ where the wave function is a product of two ordinary hydrogen-like wave functions: $\psi=\psi(1)\cdot\psi(2)$. This can be treated by Separation of Variables, and the unperturbed energy eigenvalues $E^{(0)}$ can be calculated in standard hydrogen-like fashion.
The repulsion term, which doesn't exist in the hydrogen-like atom, is treated as a perturbation: $$\hat{H}'=\bbox[yellow]{\frac{e^2}{4\pi\varepsilon_0r_{12}}}\quad.$$ As usual in perturbation theory, the first-order energy correction is calculated by sandwiching the perturbation Hamiltonian between the unperturbed wave function and its complex conjugate and integrating over all space: $$\Delta E=\iint\psi^{\ast}\frac{e^2}{4\pi\varepsilon_0r_{12}}\psi\,{\rm d}V_1\,{\rm d}V_2\quad,$$ where the double integral is over all three spherical co-ordinates for each of the two electrons. The unperturbed wave function is one describing the states of both electrons, e.g. $\psi_{1s^2}=R_{1s}(r_1)\cdot R_{1s}(r_2)$.
As long as the atom is in the ground state, both electrons individually have the same wave function, $R_{1s}(r)$, so it is impossible to distinguish them. Swapping the two electrons cannot therefore make any difference. If the atom is excited, this is usually not the case (unless both electrons are simultaneously excited into the same state, which is unlikely).
For an excited atom with one electron in the ground state and one in an excited state, the individual wave functions of the electrons are $\psi_{1s}=R_{1s}(r_1)$ and $\psi_{nl}=R_{nl}(r_2)Y_{lm}(\vartheta_2,\varphi_2)$, respectively. The unperturbed combined wave function (neglecting the repulsive term from the interaction of the two electrons) can either be $$\psi=\psi_{1s}(1)\cdot\psi_{nl}(2)\quad, \textrm{or}$$ $$\psi=\psi_{1s}(2)\cdot\psi_{nl}(1)$$ depending on which electron has been excited. Whether electron 1 has been excited or electron 2, the energy must be the same because of the symmetry - this is known as exchange degeneracy.
As in the case of the ground state, we need to use perturbation theory to find the energy correction due to the repulsive Coulomb term. Once more, the perturbation is $H'\propto\frac{1}{r_{12}}$. The difference is that now there are two degenerate wave functions. To deal with both simultaneously, we combine them into two mixed states: $$\psi_{\pm}=\psi_{1s}(1)\psi_{nl}(2)\pm\psi_{1s}(2)\psi_{nl}(1)\quad.$$ These linear combinations are then combined with the complex conjugate of one of the original wave functions when the perturbation integral is solved:
$$\Delta E\propto\iint\bbox[lightblue]{\psi^{\ast}_{1s}(1)\psi^{\ast}_{nl}(2)}\frac{1}{r_{12}}\bbox[pink]{\left(\psi_{1s}(1)\psi_{nl}(2)\pm\psi_{1s}(2)\psi_{nl}(1)\right)}\,{\rm d}V_1\,{\rm d}V_2\quad.$$
This use of linear combinations on one side of the perturbation Hamiltonian is a common adaptation of perturbation theory when dealing with degenerate states. It avoids a situation where terms in the derivation of perturbation theory cancel due to the fact that the energies corresponding to two separate wave functions are identical.
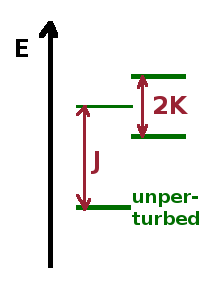
Since the perturbation Hamiltonian is just a simple factor, it commutes (i.e. can be swapped) with the functions it acts on, so we can multiply $\bbox[lightblue]{\psi^{\ast}_{1s}(1)\psi^{\ast}_{nl}(2)}$ with $\bbox[pink]{\left(\psi_{1s}(1)\psi_{nl}(2)\pm\psi_{1s}(2)\psi_{nl}(1)\right)}$ and split the integral into two parts, one containing the products of functions of the same electron and the other containing the cross terms: $$J\propto\iint\left|\psi_{1s}(1)\right|^2\frac{1}{r_{12}}\left|\psi_{nl}(2)\right|^2\,{\rm d}V_1\,{\rm d}V_2\quad,$$ $$K\propto\pm\iint\psi_{1s}^{\ast}(1)\psi_{nl}^{\ast}(2)\frac{1}{r_{12}}\psi_{1s}(2)\psi_{nl}(1)\,{\rm d}V_1\,{\rm d}V_2\quad.$$ The two integrals are known as the direct integral, $J$, and the exchange integral, $K$. Together, they determine the correction due to the perturbation caused by the second electron of the energy of the first electron and vice versa. The direct integral, due to the mutual repulsion of the two electrons, causes a shift of the energy relative to the unperturbed energy of the corresponding $\psi_{1s}\psi_{nl}$ state in a hydrogen-like atom with the same atomic number $Z$.
This may seem a little counterintuitive: how can there be a two-electron state in a hydrogen-like atom, which after all has only one electron? -- Remember the states are just solutions of the Schrödinger equation and exist even if there is no electron to occupy them!
In addition, the exchange integral causes a split in the energy of the two-electron state, i.e. there are two sub-levels separated by an energy amounting to twice the exchange integral. This is caused by the interference of the two contributing wave functions. These two distinct states are known as the symmetric solution, $\psi^S=\psi_{1s}(1)\psi_{nl}(2)\color{red}{+}\psi_{1s}(2)\psi_{nl}(1)$, and the antisymmetric solution, $\psi^A=\psi_{1s}(1)\psi_{nl}(2)\color{red}{-}\psi_{1s}(2)\psi_{nl}(1)$, respectively as they are (anti-)symmetric with respect to swapping the two electrons.
The Pauli principle determines that each state can only be occupied by a single electron. Each of the space functions for helium described on this page is therefore paired with a spin function to distinguish spin-up $(m_s=+\frac{1}{2})$ from spin-down $(m_s=-\frac{1}{2})$ states with otherwise identical quantum numbers, i.e. with the same space function: $$\psi_{n,l,m_l,s,m_s}=\psi^{space}_{n,l,m_l}\cdot\psi^{spin}_{s,m_s}\quad.$$ If each electron is to be in a distinct state, then swapping two electrons must result in a change to the wave function, i.e. the overall state must be anti-symmetric. We have seen that the space function can be either symmetric or anti-symmetric depending on the exchange integral. In order for the total wave function to be anti-symmetric, we have to pair a symmetric space function with an anti-symmetric spin function and vice versa: $$\psi_{total}=\begin{cases}\psi_{space}^A\psi_{spin}^S\\\psi_{space}^S\psi_{spin}^A\end{cases}$$
| spin function | symmetry | $S=\sum s$ | $M_s=\sum m_s$ |
|---|---|---|---|
| $\psi_{\uparrow\uparrow}$ | symm. | 1 | +1 |
| $\psi_{\downarrow\downarrow}$ | symm. | 1 | -1 |
| $2^{-\frac{1}{2}}(\psi_{\uparrow\downarrow}+\psi_{\downarrow\uparrow})$ | symm. | 1 | 0 |
| $2^{-\frac{1}{2}}(\psi_{\uparrow\downarrow}-\psi_{\downarrow\uparrow})$ | anti | 0 | 0 |
In the case of two electrons, both can be in the spin-up state ($m_s=+\frac{1}{2}$), or both spin-down, or one of each. If both spins point in the same direction, the resulting spin function is symmetric with respect to swapping the two electrons. If the two spins are different, we can either add or subtract the two spin functions, resulting in a symmetric (added) and an anti-symmetric (subtracted) combination, as shown in the table. By adding the spin quantum numbers ($s=\frac{1}{2}$ for each electron) and magnetic spin quantum numbers ($m_s=\pm\frac{1}{2}$ for each electron), we can determine the total spin, $S$, and total magnetic spin quantum number, $M_s$ for each combined two-electron state. The symmetric functions on the one hand and the anti-symmetric one on the other differ in $S$, and the different symmetric spin functions are distinguished by their $M_s$ value.
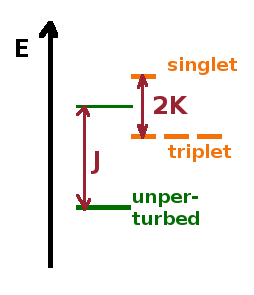
The three symmetric states together form a triplet, the anti-symmetric state on its own is a singlet. As in the case of the hydrogen-like atom previously, the conventional labels for these states include the total orbital angular momentum quantum number $L$ (expressed as a letter: s,p,d,f...) and the spin multiplicity: $^{2S+1}L$. The constituent states of the triplet are normally degenerate but, as their magnetic spin quantum number is different, they can be split if placed in a magnetic field. The quantum number for total orbital angular momentum determines the space function of the state, the spin multiplicity is determined by the spin function.
As an example, consider helium with one electron excited into the 2s state (and the other left in the 1s ground state). Since both electrons are in 's'-states ($l=0$), the total orbital angular momentum is $L=\sum l=0$, i.e. an 'S'-state in spectroscopic notation. For the symmetric spin functions, the individual spins ($s=\frac{1}{2}$) add up, so $S=\sum s=1$, while they cancel out for the anti-symmetric spin functions, i.e. $S=\sum s=0$. The symmetric spin states produce a triplet, $^3S$, while the anti-symmetric spin function results in a singlet, $^1S$.
The selection rule for transitions involving spin functions is simple: dipole radiation doesn't interact with the spin, so the spin function doesn't change: $\Delta S=0$.
The step from one electron to two electrons is quite significant because the centre of gravity of the atom is no longer static in time and repulsive interactions between the electrons need to be taken into account. The step from helium to the rest of the periodic table is comparatively simple - we just need to apply the same concepts to yet more electrons. This requires some approximations.